英茂盛业生物科技有限公司
023-67630383
023-67630383
 销售咨询QQ2548969917 技术咨询QQ1291769782
销售咨询QQ2548969917 技术咨询QQ1291769782热线/微信:13681365274
销售咨询QQ2548969917 技术咨询QQ1291769782微信扫一扫
下载说明书


长非编码RNA(lncRNA)是一类调控RNA分子,长度超过200个核苷酸,可用作新的潜在生物标志物,但其检测方法如qRT-PCR仍未得到验证,RNA降解对lnc RNA定量的影响尚不清楚。在这项研究中,测试了市售的cDNA合成试剂盒,并比较了RNA降解的影响。
使用RNA作为生物标志物的想法并不新鲜[1,2]。长非编码RNA是长度超过200个核苷酸的分子。它们被积极转录,不编码任何类型的蛋白质。关于lncRNAs的知识仍然有限,因此这是进一步研究的重要领域。lncRNA分子具有许多功能域,如RNA或DNA结合位点和蛋白质结合位点。它们也可能经历构象转换。由于这些结构域,lncRNAs可能具有重要的生理功能,如控制转录、转录后过程或翻译,或模拟表观遗传修饰。lncRNAs参与细胞过程,如增殖、凋亡、应激反应和细胞代谢或表型的调节。lncRNA表达障碍与癌症进程相关,越来越多的研究将lncRNA作为新的生物标志物[3]。
使用分子生物学方法可以很容易地检测来自组织、尿液、外周血、血清、唾液或尿液样品的lncRNA表达[4–9]。也有报道称,lncRNAs可以从外泌体中提取,外泌体被认为是细胞间通讯的介体[10]。然而,并非所有的lncRNAs都存在于每种类型的生物材料中。例如,Tang等人观察到,HOTAIR、、MALAT1、MEG-3、NEAT-1和UCA1存在于来自(口腔鳞状细胞癌)患者的癌和邻近的非癌样品中,但是在一些唾液样品中仅检测到来自这组lncRNAs的HOTAIR和malat 1[8]。
有许多可用的方法来研究lnc RNA:I)lnc RNA免疫沉淀;ii) lncRNA原位杂交;iii) Au-NP分析(基于金纳米颗粒);iv) lncRNA northern印迹分析;v)使用HRM分析(高分辨率解链)估计甲基化状态;vi)微阵列或RNA测序;和vii) qRT-PCR或新开发的ddPCR(液滴数字PCR) [5,6,11–13]。lncRNA研究中最常用的方法是杂交分析,尤其是基于SYBR-green染料和TaqMan探针的qRT-PCR方法[14]。qRT-PCR实验可以是实验室特定的。不同的研究小组使用不同的引物、参考基因(或基因组)和扩增策略,这有时使得不可能比较结果[15,16]。似乎只有商业qRT-PCR平台可以帮助解决这个问题。商业上可获得的qRT-PCR lncRNA平台,如LncProfiler qPCR Array Kit (SBI),基于Ct(阈值循环)分析,提供了在一次运行中简单快速定量90个lncRNA的可能性。与qRT-PCR相比,微阵列方法是一种经过充分验证的技术,在实验中更具可比性,但它更昂贵[17]。此外,使用商业lncRNA微阵列平台,可以评估超过30,000个lncRNA的表达,而无需使用NGS(下一代测序)数据提取所需的复杂的生物信息学方法[18,19]。然而,在qRT-PCR和微阵列方法中,检测仅限于已知的lncRNA转录物。
如上所述,在lncRNA研究中最常用的方法之一是qRT-PCR,但是这种方法没有标准化,并且使用不同的方法来制备cDNA。此外,RNA降解对lncRNA定量的影响还不清楚。在将lncRNAs作为一类新的生物标志物应用于临床之前,这些问题必须得到解决。
在这项研究中,评估了不同的cDNA合成试剂盒,并比较了RNA样品的质量。试剂盒基于以下内容:I)随机六聚体引物,之前有聚腺苷酸加尾和接头锚定步骤;ii)使用随机六聚体引物和oligo(dT)的混合物的简单反应;iii)只有随机六聚体引物;和iv)仅oligo(dT)。
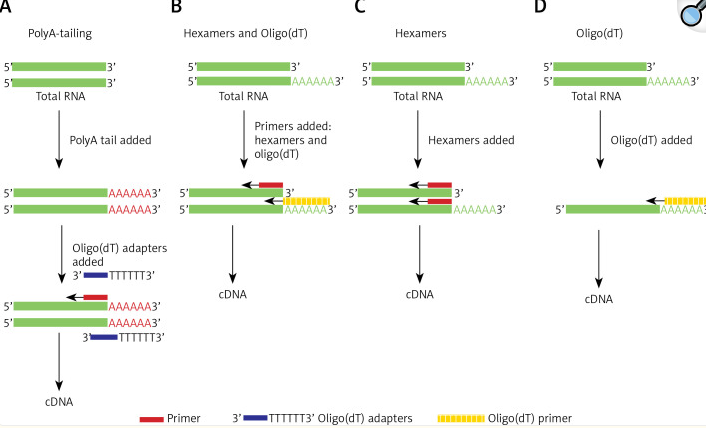
在这项研究中,不同的cDNA合成试剂盒基于:I)随机六聚体引物,之前有聚腺苷酸加尾和接头锚定步骤;ii)使用随机六聚体引物和oligo(dT)的混合物的简单反应;iii)仅使用随机六聚体引物;和iv)仅测试寡(dT)。我们假设Ct值越低,cDNA合成的方法就越灵敏和精确。平均qRT-PCR Ct值的比较显示在热图上(图2 A)。观察到61/90 (67.78%)受检lncRNAs的Ct值较低,其中cDNA是使用随机六聚体引物合成的,之前有聚腺苷酸加尾和接头锚定步骤。然而,在15/90(16.67%)lnc RNA的情况下,观察到其他方法的Ct值更高(图2 A和B)。
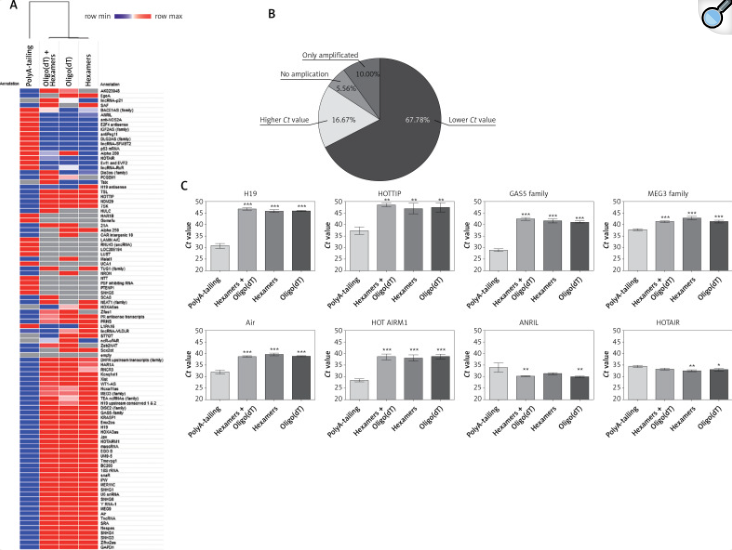
热图和聚类显示,使用随机六聚体引物获得的cDNA的qRT-PCR Ct值与其他检测的cDNA合成试剂盒不同,这些试剂盒彼此相似并聚类在一起(图2 A)。此外,9/90 (10%)被检测的lncRNAs仅在通过随机六聚体引物获得的cDNA样品中检测到,所述随机六聚体引物之前有聚腺苷酸加尾和接头锚定步骤。然而,对于5/90 (5.56%)的lncRNA,没有观察到lnc RNA扩增,并且在使用随机六聚体引物和oligo(dT)的组合以及仅使用随机六聚体引物或仅使用oligo(dT)合成的cDNA样品中检测到这些lnc RNA(图2 A和B)。
对于癌症中最常描述的lncRNAs,特别是头颈部鳞状细胞癌[3],在使用不同cDNA合成试剂盒获得的qRT-PCR Ct值中观察到显著变化。使用先加尾再随机六聚体引物转录获得的cDNA,对于H19 (p < 0.0001)、HOTTIP (p = 0.0011)、GAS5家族(p < 0.0001)、MEG3家族(p = 0.0001)、Air (p < 0.0001)和HOTAIRM1 (p < 0.0001),观察到较低的Ct值(32.55±4.12对42.49±3.52),对于ANRIL (p = 0.0045)和HOTAIR (p = 0.0053)有更高的Ct值(34.18±0.33对31.68±1.46)(图2 C)。
然后研究了总RNA降解对qRT-PCR定量的影响。新鲜分离的RNA被等分(V = 5μl/样品,C = 1μg/样品),并在室温下孵育3、6、8和10天。观察到总RNA的降解(图3 A)。孵育3天后,没有观察到28S条带,但观察到18S rRNA条带的涂片,直到第10天,此时RNA高度降解(没有28S或18S rRNA条带;图3 A)。降解过程导致培养10天后RNA样品吸光度的变化,使用纳米滴分光光度计测量该变化。紫外吸光度增加,并被视为表观浓度变化的结果,从分离后立即测得的206.3纳克/微升到培养10天后测得的235.27纳克/微升(p = 0.0227图3 B)。
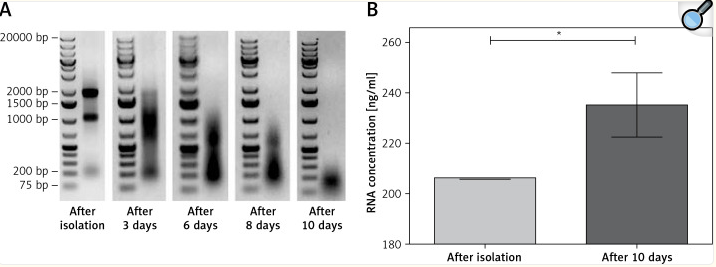
使用随机六聚体引物进行cDNA合成,之前进行聚腺苷酸加尾和接头锚定步骤,并使用LncProfiler qPCR阵列试剂盒进行qRT-PCR,用于从分离当天以及孵育3天和10天后的RNA样品。对于83% (75/90)的lncRNAs,高质量RNA和降解RNA(第3天或第10天)的Ct值没有显著差异(p > 0.05)。对于lnc RNA:Jpx、PSF抑制RNA、PTENP1、SAF、SNHG5和mRNA GAPDH,在分离后或温育3天后使用RNA样品合成的cDNA的Ct值之间没有差异,但是当基于温育10天的RNA进行qRT-PCR时,观察到样品的Ct值显著更高。
结论:
与大多数研究人员类似,我们也选择了qRT-PCR方法来研究lncRNA表达,因为它简单且样品分析快速[20]。然而,qRT-PCR的主要问题是在实验工作流程中选择合适的方法,但是没有实验数据来比较针对该问题的各种方法。在这项研究中,我们比较了三种不同的市售cDNA合成试剂盒。基于总RNA(包括lncRNA部分),我们获得了cDNA,这可用于使用商业上可获得的LncProfiler qPCR阵列试剂盒(SBI)定量lncRNA的表达。我们假设Ct值越低,该方法越灵敏和精确。比较三种不同的方法,我们观察到使用cDNA合成的方法的最佳结果(低Ct值),该方法通过随机六聚体引物,在此之前进行聚腺苷酸加尾和接头锚定步骤。一般来说,使用随机六聚体引物和oligo(dT)的混合物或仅使用随机六聚体引物或oligo(dT)的简单反应的结果可能会错误地提示一些lncRNAs没有表达或表达水平非常低。值得注意的是,一些lncRNAs具有内源性polyA尾,而其他的则没有。此外,大多数lncRNAs以低拷贝数存在于样品中,这给它们的定量造成困难[21,22]。这两个lncRNA特征需要使用cDNA试剂盒,并额外添加polyA尾和退火锚(dT)接头。我们观察到,这种方法可以提高lncRNA定量的特异性和敏感性。不幸的是,在大多数研究中,使用包含oligo(dT)和/或随机六聚体引物的混合物的试剂盒制备cDNA[14,23,24],其中oligo(dT)对没有polyA尾的lncRNA分子不起作用。众所周知,特异性引物会减少背景引发,但随机引物和寡(dT)引物会最大限度地增加可从少量RNA样品中分析的mRNA分子数量[25]。我们观察到,人工添加polyA尾,然后使用退火锚(dT)接头(寡(dT)引物的类似物)可以帮助更精确地定量lncRNAs。
对我们观察到的一些lncRNAs定量困难的一个可能的解释是分子结构特征的影响,如长度、GC含量和分子折叠。众所周知,短RNA分子如miRNAs很难通过qRT-PCR定量,需要添加接头或polyA尾的特异性cDNA合成试剂盒。较长的分子似乎比短的RNA(如miRNAs)更容易定量。另一方面,许多二级结构如发夹环对cDNA合成是有问题的,需要更高的温度来松弛RNA分子,使它们可用于逆转录酶[26,27]。此外,我们使用的一种试剂盒具有DTT(二硫苏糖醇)增强子,这有助于扩增富含GC序列的模板,并有助于获得更好的qRT-PCR结果[28]。由于结构特征,一些lncRNAs似乎需要在cDNA合成过程中添加polyA尾或更长时间的更高温度。我们认为一些结果可能被忽略,因为使用了不适当的cDNA合成方法,并且没有对特定的lncRNA进行定量。
下一个问题是lncRNAs的稳定性和RNA完整性对qRT-PCR定量的影响。由于lncRNAs的长度(超过200 nt),与miRNAs等短RNA分子相比,它们被认为不太稳定,更容易降解。我们使用cDNA试剂盒分析了RNA稳定性对lncRNAs定量的影响(用锚定(dT)接头退火的polyA加尾和使用随机引物混合物的cDNA合成),我们比较了高质量总RNA(可见28S和18S rRNA带)、降解(无28S带和可见18S rRNA带涂片)和高度降解RNA(无28S或18S rRNA带)的样品。降解导致RNA结构的变化,增加短RNA分子的数量,这强烈影响RNA检测[29,30]。我们的结果表明,大多数检测的lncRNAs是稳定的,即使当总RNA高度降解时。我们的观察结果得到了Kraus等人的支持。他们报道说,一些lncRNAs比大约22个核苷酸长的miRNAs更稳定。
然而,一些长转录物,如mrna,对降解很敏感,在这种情况下,RNA完整性是影响qRT-PCR定量的重要因素。据推测,lncRNA半衰期取决于其在基因组中的编码位置、转录后修饰以及亚细胞定位及其功能[33]。一些作者注意到lnc RNA在基因组中的定位可能会影响其转录稳定性,特别是与那些来自内含子的lnc RNA(半衰期超过16小时)相比,基因内和顺式反义lnc RNA[8,33]。我们注意到一些lncRNAs稳定的时间更长。3天后,我们在大多数被检查的lnc RNA中没有观察到Ct值的变化,而10天后,仅在一些lnc RNA中观察到Ct值的变化。唾液中一些lncRNAs的检测证实了这些分子的高稳定性[8]。此外,从血浆中分离的lncRNAs对RNase A消化和室温下过夜孵育具有抗性[9]。一般来说,低RNA稳定性可能会给分析从存档的福尔马林固定石蜡包埋(FFPE)块中获得的一些长编码或非编码RNA转录本带来困难[34]。这个问题通过应用通过与三对不同的非重叠引物反应来测量lncRNA表达水平的简单修改来解决[35]。然而,在FFPE区块的存档过程中,RNA被降解和修饰,这使得该RNA更难以分析。
众所周知,理想生物标志物分子的主要特征是采集简单、来源多样以及测量方法简单[20,37]。我们假设在适当的加工条件下,lncRNA似乎符合生物标记分子的特征。然而,在临床使用lncRNA生物标志物之前,应该应用标准化程序。
总之,我们建议如下:I)使用设计用于lncRNA或cDNA合成的cDNA试剂盒,在此之前用锚定(dT)接头退火的polyA加尾;ii)使用设计用于高温下长时间反应的高度热稳定的逆转录酶;和iii)通常推荐使用高质量的RNA,但是大多数lncRNAs似乎是稳定的,并且可以在降解的RNA样品中定量。

