英茂盛业生物科技有限公司
023-67630383
023-67630383
 销售咨询QQ2548969917 技术咨询QQ1291769782
销售咨询QQ2548969917 技术咨询QQ1291769782热线/微信:13681365274
销售咨询QQ2548969917 技术咨询QQ1291769782微信扫一扫
下载说明书


https://doi.org/10.1080/15476286.2021.1899500
长非编码RNA(lnc RNA)由于其在多种生物过程和疾病中的意义而日益成为研究的焦点。然而,大多数lncRNAs丰度低,保守性差,给功能研究带来了挑战。CRISPR/Cas系统是过去十年中出现的一项创新技术,可以用来进一步了解lncRNA的功能。该系统通过指导RNA (gRNA)和Cas核酸酶复合物靶向特定的DNA和/或RNA序列。我们和其他人已经在各种应用中利用这种技术,如lncRNA敲除、敲除、过表达和成像。在这篇综述中,我们总结了CRISPR/Cas技术如何提供新的工具来研究lncRNAs的作用和治疗意义。
简介
由于只有大约2%的人类基因组含有蛋白质编码基因,人们对人类基因组非编码区的作用越来越感兴趣。使用最新的测序技术,已经鉴定出各种调节性非编码RNA。这些非编码RNA(ncRNAs)包括长度< 200核苷酸(nts)的小NC RNAs,如miRNAs和piRNAs,以及长度> 200 nts的长非编码RNA(lnc RNAs)[citation 2]。尽管低水平表达,保守性差,并表现出组织特异性表达[Citation2,Citation3],但lncRNAs已被证明在多种细胞过程中发挥重要作用,如控制转录,充当分子诱饵,指导定位,并提供蛋白质支架[Citation3]。lncRNAs可定位于细胞核和/或细胞质,对DNA、RNA和/或蛋白质产生影响。由于低丰度,组织特异性表达,广泛的细胞作用和有限的研究技术,研究lncRNAs可能是复杂和具有挑战性的。
通常,为了研究一种新的lncRNA的功能,需要诱导其表达发生变化。然而,广泛使用的工具,如RNA干扰(RNAi)和反义寡核苷酸(ASOs),在lncRNA研究中效果不佳[引文4–6]。RNAi使用多蛋白RNA诱导沉默复合物(RISC)来诱导由小干扰RNA (siRNA)结合靶RNA介导的RNA降解[Citation7]。尽管RNAi在细胞质分子的研究中有用,但由于RISC在大多数细胞类型中主要定位于细胞质,RNAi在抑制细胞核lncRNA分子方面通常不太有效。ASOs的功能是与RNA结合,然后通过识别DNA-RNA杂合体的RNase H1酶选择性降解ASO-RNA杂合体。ASOs的使用依赖于外源性修饰的寡核苷酸来产生瞬时效应,这使得该技术变得复杂。这些技术的最大限制是lncRNAs的降解,这限制了它们在作用机制研究中的应用,同时不会产生功能的完全丧失。
从21世纪初开始,基因组操作技术的广泛使用,如锌指核酸酶(ZFNs),改变了分子生物学的范围。不久之后,转录激活因子样效应核酸酶(TALENs)的使用随之而来。近20年后,这些技术仍然流行,现在已被用于lncRNA研究。例如,Lee等人(2016)能够使用TALENs技术,利用同源定向插入,将含有串联聚腺苷酸化信号的Lox-Stop-Lox盒插入NORAD基因座,沉默lncRNA的表达[Citation8]。虽然这些系统已经完成了lncRNAs的高级遗传操作,但它们仍然存在时间、成本和灵活性方面的问题,以及蛋白质工程、合成和验证方面的挑战[引文9]。
对lncRNAs进行遗传操作的更有效的方法是成簇的规则散布的短回文重复序列(CRISPR)系统。CRISPR系统是一种基因组编辑技术,由于其易用性和灵活性,它已成为分子生物学中的一种革命性工具。因为它主要依赖于使用gRNAs (gRNAs)来指导Cas酶的催化活性[Citation10,Citation11],所以它优于更费力的ZFNs或TALENs。Cas酶产生双链断裂(DSB),通常通过非同源末端连接(NHEJ)来修复。虽然CRISPR系统传统上使用Cas9,但它也可以与dCas9、Cas12和Cas13等其他酶一起使用,从而提供极大的灵活性。催化死亡的Cas9 (dCas9)通过阻断转录来破坏基因功能。Cas12有两个功能已知的直向同源物;Cas12a靶向单链DNA (ssDNA) [Citation12],而Cas12b靶向双链DNA [Citation13]。Cas13选择性靶向单链RNA (ssRNA),并通过RNA降解被用于编辑RNA、成像RNA和敲除转录物[Citation14,Citation15]。
由于其多功能性和灵活性,CRISPR系统在lncRNAs的研究中特别有用。该技术能够靶向细胞核或细胞质lncRNA,破坏基因组以选择性地与(或避免)邻近的遗传物质相互作用,并上调或下调转录,所有这些都不会降解lnc RNA,这为高级作用机制研究创造了条件。各种Cas酶的适应性为靶向基因位点和RNA转录物提供了机会。通过这种灵活性,研究可以更好地解决lncRNAs的高度特异性和多方面的作用。虽然这些技术中有许多是最近发展起来的,但如果选择得当,CRISPR/Cas系统可以极大地促进我们对lncRNA功能的理解。在这篇综述中,我们强调了CRISPR/Cas系统在研究lncRNA功能中的各种用途。
1. 用Cas9靶向lncRNA基因
CRISPR/Cas9最基本的用途是利用gRNA通过DSB和NHEJ诱导无义或移码突变。虽然这是一种破坏蛋白质编码基因正常蛋白质生产的有效方法,但这些突变很少显著到足以使lncRNA基因失活。由于进化保守性差,通常不清楚lncRNA的哪个区域是活性结构域,这阻止了gRNA靶的有效选择。如果能够选择lncRNA基因上的靶位点,重要的是要考虑脱靶效应以及对邻近基因的影响。lncRNAs可能是基因间或基因内的。基因内lncRNA可能与蛋白质编码基因重叠,或者可能位于宿主基因的内含子内(内含子lnc RNA)。lncRNA启动子可能存在于相邻基因中,或者与附近的蛋白质编码基因共享[Citation16]。lncRNA启动子在基因组中的跨度对CRISPR敲除系统的使用构成了重大限制,因为靶向lncRNA可能对附近的基因产生重大影响[Citation16]。由于这些限制,CRISPR系统的新用途已经出现。表1总结了其中的一些工具。
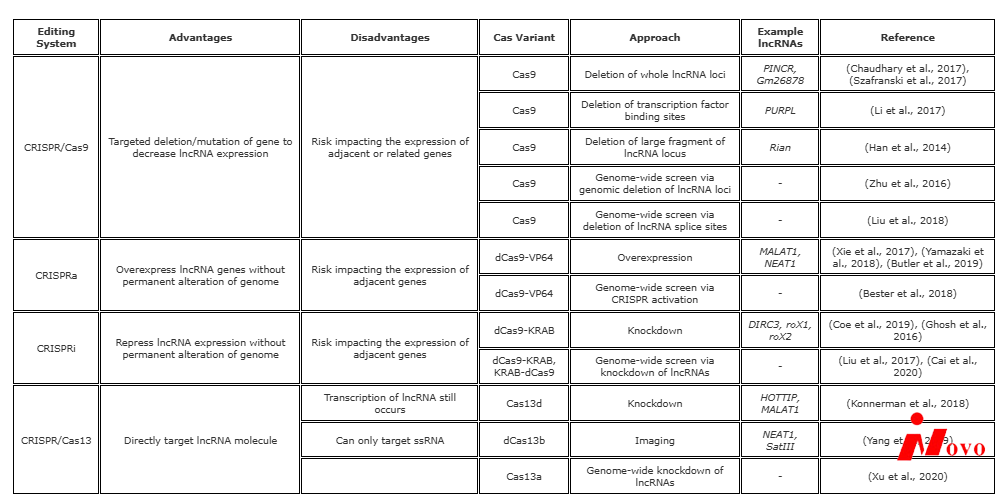
1.1使用Cas9进行基因编辑
产生有效lncRNA敲除的一种方法是使用两对gRNAs,靶向lncRNA基因的起始和末端,切除完整的lncRNA基因座。已经发现,引入多个gRNAs增加了这种敲除的效率[引用17,引用18]。许多研究试图切除lncRNA基因座的大片段,而不是完整的lnc RNA[引文17,引文19,引文20]。例如,Han等人(2014年)删除了高达23 kb的Rian,一个57.8 kb的lncRNA基因,使用Cas9介导的删除成功降低了雌性小鼠后代中的Rian表达[Citation17]。Zhu等人(2016)使用这种技术进行高通量筛选,筛选了超过700种具有致癌或肿瘤抑制活性的lncRNAs引文19]。他们还发现,当这两种gRNA被导入单个慢病毒载体的两个独立的U6启动子下时,配对的gRNA系统更有效[Citation19]。一些研究已经使用这种技术成功地删除了完整的lncRNA基因座[Citation5,Citation21,Citation22]。
或者,Cas9可以靶向lncRNA的转录因子结合位点或启动子。有人提出,lncRNA转录过程中转录因子的募集可能会影响附近基因的转录[Citation23]。通过突变lncRNA的启动子,可以阐明lncRNA转录对邻近基因的影响,而不是lncRNA转录本身对邻近基因的影响[引文24]。Engreitz等人(2016)进行了一项研究,他们使用2-3个gRNAs成功敲除了12个lncRNAs的启动子,以检测对相邻基因表达的影响[Citation25]。他们发现在12个lncRNA中的5个中,敲除ln crna启动子影响了靶区域大约5-71kb的基因[引文25]。一种类似的方法已被用于确定lncRNAs的作用,包括UCA1、P14AS和PURPL,通过敲除其启动子中的转录因子结合位点[引文26-28]。这种类型的敲除已被发现非常有效,Zhen等人(2017)证明了这一点,他们发现靶向UCA1启动子的gRNA使UCA1表达减少了80%[citation 26]。当敲除P14AS上游的启动子样序列时,Ma等人(2020)发现P14AS转录减少以及下游ANRIL表达减少[引文27]。
研究lncRNA的另一个目标位置是ln crna基因内的剪接位点。以前已经证明用ASOs靶向剪接位点能有效敲低lncRNA表达[Citation29]。同时,CRISPR/Cas9系统已被用于靶向DNA序列上的前mRNA剪接位点,通过无义介导的衰变导致选择性剪接和基因敲除[Citation30,Citation31]。通过结合这些技术,gRNAs可以靶向Cas9到lncRNA剪接位点,以产生有效的内含子保留或外显子缺失[Citation32]。Liu等人(2018)在全基因组范围内使用这种技术来筛选必需的lnc RNA[citation 32]。值得注意的是,后来的研究表明,使用这种方法的定向缺失可能不如以前建议的有效[引文33]。用CRISPR/Cas9靶向lncRNA剪接位点既可用于功能丧失研究,也可用于lncRNA筛选。虽然这种方法并不常见,但它提供了一种解决方案,可以删除大部分DNA,同时产生有效的敲除,使脱靶效应最小化。
最后,敲入模型可以用来研究lncRNAs在细胞中的功能。由于lncRNA的大尺寸,敲入lnc RNA是不实际的。研究使用CRISPR介导的内源性lncRNA追踪(CERTIS)系统,将标签敲入lncRNA基因,以可视化lncRNA定位[Citation34]。或者,Lee等人(2016年)利用TALENS敲入一个转录终止元件,使NORAD基因座失活[Citation8]。Yin等人(2015)敲入polyA终止盒,使用CRISPR/Cas9 [Citation18]使Haunt1 lncRNA转录物减少55%。
1.2使用Cas9进行基因编辑的挑战
使用两个gRNAs切除大片段lncRNA的主要限制是影响相邻基因的显著风险,例如那些可能与lncRNA基因重叠的基因,那些受lncRNA转录调节的基因,或那些在ln crna基因座中具有可能调节其他基因的DNA元件的基因[Citation35]。虽然将Cas9靶向转录因子结合位点、启动子或剪接位点可最大限度地降低删除其他重要遗传物质的风险,但这些技术存在自身的问题。当操作lncRNA启动子时,仍有可能影响其他基因,例如由与lncRNA相同的启动子调控的基因或可能与靶向启动子重叠的基因。关于敲入,这些类型的研究需要广泛的研究,以确定潜在的脱靶效应。撞击发生的地点的选择是至关重要的。然而,对于特定的lncRNA,当决定在哪里进行敲入时,必须考虑lnc RNA可能与相邻基因的双向关系。总的来说,对编码lncRNA的DNA的操作提出了关于破坏周围遗传物质的问题,这是由于ln crna的大尺寸以及它们与其他基因组元件的重叠和相互作用。
1.3沉默或过表达
CRISPR干扰(CRISPRi)工具提供了一种抑制基因表达而不永久突变基因组的方法,而不是像上一节所述的那样在敲除或敲入模型中永久改变DNA。CRISPRi利用dCas9和gRNA阻断RNA聚合酶II转录,可逆地抑制感兴趣基因的功能活性[Citation36,Citation37]。这项技术已经用于lncRNA筛选[引用38,引用39]以及沉默研究[引用40–42]。为了进一步增强基因阻遏,转录阻遏物如KRAB可以融合到dCas9-gRNA系统中[Citation43]。尽管在CRISPRi研究中广泛使用KRAB,但人们发现,在lncRNAs的CRISPRi研究中,替代转录阻遏物,如源自MXD1蛋白的SIN3相互作用结构域,可能比KRAB更有效[Citation44]。
在CRISPRi发明后,利用融合到RNA聚合酶募集因子的dCas9开发了CRISPR激活(CRISPRa)[citation 45]。VP64、p65和Rta等转录激活剂通常用于诱导基因表达[Citation16]。CRISPRa可用于筛选和过表达研究[Citation46]。这项技术已被用于阐明lncRNAs的功能,如MALAT1和neat 1[citation 47–49]。
1.4 CRISPRi/CRISPRa系统的挑战
CRISPRi和CRISPRa有相似的优缺点。首先,由于不会破坏DNA序列或导致任何永久性的改变,这些方法不太可能永久性地影响邻近的基因。尽管如此,转录激活子和阻遏子的募集可能引起相邻基因表达水平的变化。例如,当在HEK293T细胞中过度表达lncRNA RP11-326A19.4时,Soubeyrand等人(2019)发现细胞因子IL6的表达增加,这表明CRISPRa具有不可预测的脱靶效应[Citation50]。通过利用转录阻遏物或激活物,可以观察lncRNA表达对附近基因的影响。这是很重要的,因为lncRNA本身的转录可以调节一些邻近的基因。CRISPRa系统诱导lncRNA亚型以天然剪接的比例过度表达,而不是可能只诱导特定亚型表达的敲入模型。CRISPRi和CRISPRa可能与附近的启动子相互作用,导致混淆结果,如果感兴趣的lncRNA的启动子在另一个基因内,则应该考虑这种限制。
2.用Cas13靶向lncRNA分子
最近,使用CRISPR/Cas13靶向RNA的情况有所增加。CRISPR/Cas13系统采用Cas13,一种与CRISPR RNA (crRNA)组装的RNA酶,以形成可编程的RNA靶向系统。Cas13蛋白包含两个具有内源性RNA酶活性的高等真核生物和原核生物核苷酸结合(HEPN)结构域[Citation51]。目前,有四种已知的Cas13蛋白直向同源物(Cas13a、Cas13b、Cas13c和Cas13d),每种在人类细胞系中表现出不同水平的活性[Citation52]。该系统主要用于lncRNA敲除[Citation15,Citation53]和成像系统[Citation54],尽管它有潜力用于一系列侧重于lncRNA功能的研究。
2.1用Cas13敲减IncRNA
先前敲除lncRNA的方法,如RNAi或ASOs,有可能诱导显著的脱靶效应[引文55-58]。甚至CRISPRi也能产生不可忽略的脱靶效应,在单细胞克隆中诱导无意的转录变化[Citation59]。与RNAi相比,CRISPR/Cas13方法已被证明具有相似的敲除效率,具有更少的非特异性效应[引文60]。因为Cas13可以定位于整个细胞,它有可能敲除细胞核和细胞质的lncRNAs。Konermann等人(2018年)使用CasRx(一种Cas13d直向同源物)表明,该系统可以在人类细胞系中特异性、有效地敲除lnc RNA[citation 15]。Xu等人(2020)也利用CRISPR/Cas13系统进行了高通量筛选,成功敲除了非常长的基因间非编码(vlinc)RNAs[引用53]。这一发现表明该系统可用于整个细胞中lncRNAs的表型分析。
2.2 CRISPR/cas 13系统的挑战
虽然CRISPR/Cas13系统提供了一种针对特定RNA的强大方法,但它有一些局限性。该系统利用Cas13的各种直向同源物,每种都有其敲除效率的限制。这对于依赖于直向同源物的某些模式生物的靶位置和sgRNA设计造成了限制。使用Cas13a的体外和细菌研究发现,该系统优先靶向具有原间隔区侧翼序列(PFSs)的区域[Citation61]。Cas13b系统也显示出在细菌细胞中对PFS的偏好[Citation62,Citation63],尽管在哺乳动物细胞中关于PFS需求的发现是矛盾的[Citation64]。靶lncRNA的二级结构也限制了CRISPR/Cas13系统的能力。因为Cas13只能靶向ssRNA,所以该系统有潜力在没有高阶结构的lncRNAs中发挥良好的作用[引文61]。然而,许多lncRNAs确实包含高阶或双链结构区域,使sgRNA设计变得复杂[引文64]。这个挑战可以通过研究感兴趣的lncRNA的结构来克服。Bandaru等人(2020年)开发了一个工作流程,使用结构序列数据战略性地确定lncRNA单链区域内合适的sgRNA靶标,Wessels等人(2020年)开发了一个程序,预测Cas13的gRNA靶标[Citation64,Citation65]。尽管存在挑战,但CRISPR/Cas13系统是一个强大而具体的工具,可用于敲除或可视化lncRNAs。
3.利用CRISPR对lncRNAs进行功能鉴定
通过正确选择和执行各种CRISPR技术,人们可以对lncRNAs进行超越传统敲除、敲除和敲入的高度细致的操作。
3.1 lncRNA成像
此前,MS2-衣壳蛋白(MS2-MCP)系统已被用来可视化RNA [Citation66]。然而,该系统使用的适体有可能破坏RNA结构、表达和功能[Citation54]。Wang等人(2019)开发了一个系统,名为CRISPR活细胞荧光原位杂交(CRISPR LiveFISH),其中结合荧光团的寡核苷酸与dCas9和dCas13结合使用,以可视化活细胞中的DNA和RNA[citation 67]。虽然该系统尚未在lncRNA研究中进行测试,但它为未来的研究提供了很大的希望。同时,Yang等人(2019)使用活细胞成像方法对lncRNAs进行了可视化,该方法使用荧光标记的无催化活性的Cas13 (dCas13)蛋白对细胞核和细胞质RNA进行可视化[Citation54]。通过分析功能性dCas13直向同源物,他们确定dPspCas13b和dPguCas13b是有效的dCas13蛋白,可以标记哺乳动物细胞中的lnc RNA[citation 54]。该系统被用于有效地确定核lncRNA,NEAT1的定位。还使用两种dCas13蛋白进行双色成像,以同时观察两种细胞核lncRNAs,NEAT1和SatIII。这项技术允许双重lncRNA跟踪,这有可能扩展到可视化任何细胞区室中的ln crna。该系统还可以与dCas9配对,使用双色系统同时显示DNA和RNA。
3.2确定lncRNA是顺式作用还是反式作用
在研究lncRNA的功能时,确定它在细胞内的位置是很重要的。为了确定lncRNA的细胞定位,可以如前所述进行亚细胞分离或dCas13成像[Citation54]。一旦确定了感兴趣的lncRNA的定位,就可以确定lncRNA是顺式作用还是反式作用。顺式作用lncRNA调节周围基因的表达和/或染色质状态,而反式作用lncRNA在远处调节基因表达[Citation68,Citation69]。为了确定lncRNA是否反式作用,可以在CRISPR/Cas9敲除细胞中进行拯救实验。如果拯救实验后出现敲除表型逆转,那么lncRNA可能反式作用[Citation68]。lncRNA在细胞质和/或核质中的定位进一步支持lncRNA是反式作用的[Citation69]。然而,重要的是要注意,如果lncRNA主要定位于细胞质,其编码潜力应受到质疑(见[引文70])。
由于lncRNA的功能和转录位点之间的模糊性,研究顺式作用ln crna可能会带来挑战[引文68]。然而,CRISPR/Cas系统可以提供对顺式作用lncRNA功能的深入了解。如果lncRNA的转录发生在单一启动子处,可以对lncRNA的第一个外显子内的小区域进行CRISPR/Cas9敲除,然后可以确定附近基因的表达[Citation68]。由于CRISPR/Cas9系统对基因组DNA的直接操作,这类研究出现了一个主要的混淆。产生的表型可能是由于成熟RNA的改变或lncRNA转录完全缺失。对lincRNA-p21的研究显示了这种现象的一个很好的例子。在研究了p21在lincRNA-p21/-小鼠胚胎成纤维细胞(MEFs)中的表达后,Linc RNA-p21被归类为顺式作用,因为发现它激活p21,一种邻近基因的表达[Citation71]。或者,Groff等人(2016)的一项研究表明,并不是lincRNA-p21的表达介导了其在p53信号通路中的作用,而是位于lincRNA-p21基因座内的DNA增强子元件[引用35]。为了更好地阐明lincRNA-p21的功能,可以利用CRISPRi或CRISPRa技术进一步研究顺式激活或基因座增强子区域的作用[citation 72–74]。然而,这些方法可能在染色质环境中引入无意的变化,这可能被错误地归因于lncRNA转录的改变。此外,可以使用CRISPR/Cas13敲除lncRNA,尽管lncRNA转录的结果可能无法与敲除后观察到的表型区分开来。
研究lncRNA顺式作用的另一种方法是通过将转录物与邻近基因结合。Luo等人(2016)利用dCas9和融合到感兴趣的lncRNA的gRNA来引导和限制复合物到附近的基因座,确定lncRNA是否调节靶基因的表达[Citation75]。另一种研究lncRNA功能的方法是CRISPR-Disp(CRISP-Disp)[citation 76,Citation77]。Shechner等人(2015年)设计了这种技术,将大型lncRNA货物靶向内源性位点[Citation76]。该系统使用具有整合的功能性lncRNA结构域和融合到VP64的dCas9分子的grna[citation 77]。Shechner等人(2015年)发现,复合物能够诱导内源性基因座的显著激活。该系统有可能用于其他应用,如内源性和融合蛋白募集以及亲和标记[Citation76]。dCas9栓系和CRISPR-Disp都可以用来阐明顺式作用lncRNAs的功能。
3.3确定lncRNA中的潜在结构域
确定lncRNA的结构可以阐明其潜在的功能,并有助于gRNA的设计。许多lncRNAs具有复杂的二级和三级结构,为蛋白质、RNA或DNA相互作用提供界面[Citation78]。与许多蛋白质编码基因不同,许多lncRNAs丰度低[Citation79]且保守性差[Citation80]。这对识别lncRNAs中的功能域提出了挑战。更复杂的是,RNA结合蛋白可能与lncRNA转录物上的小而保守的区域结合,使得很难区分是lncRNA还是蛋白质介导了观察到的表型。CRISPR-Disp可用于分析这些结构域。使用该系统,未表征的lncRNA结构域可以被靶向到感兴趣的位点,以研究它们的潜在作用。平铺CRISPR也可用于检测功能性lncRNA结构域。Wang等人(2019)开发了这种新的CRISPR/Cas9方法来表征Xist的功能域,Xist是一种参与X染色体失活的lnc RNA[citation 81,Citation82]。通过引入超过1500个grna,平铺整个Xist转基因,作者从重叠的grna中探测簇状突变,表明一个功能域并成功识别基因的必需区域[Citation81]。利用平铺CRISPR技术的屏幕将有可能表征lncRNAs的功能域。
3.4体内研究
确定lncRNA功能的最全面的方法之一是研究其在动物模型中的作用。传统的体内研究使用Cre-LoxP系统,其中组织特异性Cre重组酶(Cre)在两个LoxP位点之间执行基因组DNA的靶向删除[Citation83]。尽管该系统确实提供了时间和组织特异性控制,但这可能是一个昂贵且耗时的过程[Citation84]。CRISPR/Cas9系统提供了一种生物遗传修饰的替代方法。如前所述,这已经通过使用gRNAs对在鼠模型中精确删除高达23kb的lncRNA基因座实现[Citation17]。还通过CRISPR/Cas9系统在外显子内插入polyA盒来沉默lncRNA表达,以促进转录物的切割并终止转录[Citation85]。此外,使用piggyBac转座子和Cre诱导的dCas9系统的组合,CRISPRa已被开发用于在体内激活lncRNA基因座[Citation86]。
当建立体内系统来研究lncRNAs时,必须考虑非特异性gRNA靶标[引文87]。当对用CRISPR/Cas9系统编辑的小鼠进行全基因组测序时,发现了数量惊人的单核苷酸变体[Citation87]。虽然该系统可以诱导显著的脱靶突变,但最近的一项研究开发了一种方法,提供了体内脱靶(vivo)的验证[Citation88]。体内工作流程包括两个步骤:通过测序鉴定潜在脱靶切割位点的体外切割效果报告,以及通过深度测序在CRISPR/Cas9编辑的靶组织中进行脱靶突变验证[Citation88]。这种方法可以与计算机技术相结合,后者设计的grna具有最少数量的相似基因组位点,以评估grna的准确性[引文88]。这项新兴技术显示了我们未来通过最小化潜在脱靶效应在动物模型中进行lncRNAs功能研究的能力。
3.5 CRISPR库
CRISPR文库筛选将gRNAs池引入细胞群体,敲除(如在Cas9的传统情况下发生的)各种基因,以识别基因如何在对选择压力的响应中富集或消耗。CRISPR文库筛选可以在体外和体内进行,使用成对的grna来敲除lncRNAs或单个grna来靶向剪接位点[Citation19,Citation32,Citation89,Citation90]。屏幕还利用了CRISPRi和CRISPRa系统[Citation89,Citation91]。这些研究为在无偏见和全基因组范围内确定新的lncRNA作用和相互作用提供了机会,阐明了细胞质和细胞核的相互作用。然而,全基因组lncRNA筛选可能很困难,因为ln crna的大小和点突变不能产生与蛋白质编码基因筛选相同的失活。
结论
尽管低表达[Citation79]和保守性差[Citation80],CRISPR/Cas系统通过敲除、敲除、过表达和成像方法提供了研究lncRNAs潜在作用的创新方法(图1)。除了这篇综述中提到的许多技术之外,各种其他的进展也大大增加了我们对RNA和lncRNAs的了解。无催化活性的CasRx (dCasRx)系统已被用于RNA剪接的操作[Citation15]。因为lncRNAs经历类似于mRNAs [Citation92]的剪接,该系统可用于对这些转录物进行剪接异构体工程。此外,dCas13与作用于RNA 2型(ADAR2)酶的腺苷脱氨酶融合后,可有效编辑RNA[citation 63]。该系统被称为用于可编程A到I替换(修复)的RNA编辑,不受序列限制,并具有支持lncRNA编辑的潜力。一个类似的名为RESCUE(针对特定C-U交换的RNA编辑)的系统扩展了修复系统,允许A到I和C到U的RNA编辑[Citation93]。CRISPR/Cas13系统还被设计为通过特定的高灵敏度酶促报告解锁(夏洛克)[Citation94]等方法进行敏感的RNA检测。由于系统的可编程性质,这些检测方法可能用于lncRNA研究。高等人。al (2021)能够操纵CRISPR系统,使用带有CRISPR激活子相关报告基因的内源性转录门控开关,可靠地量化低拷贝数lncRNA表达水平[Citation95]。CRISPR/Cas9技术也可用于通过利用与乙酰转移酶催化结构域融合的dCas9蛋白来操纵lncRNA基因的表观遗传调节[Citation96]。这将允许各种lncRNA基因座的受控反式激活,从而提供了另一种询问lncRNA功能的系统方法。
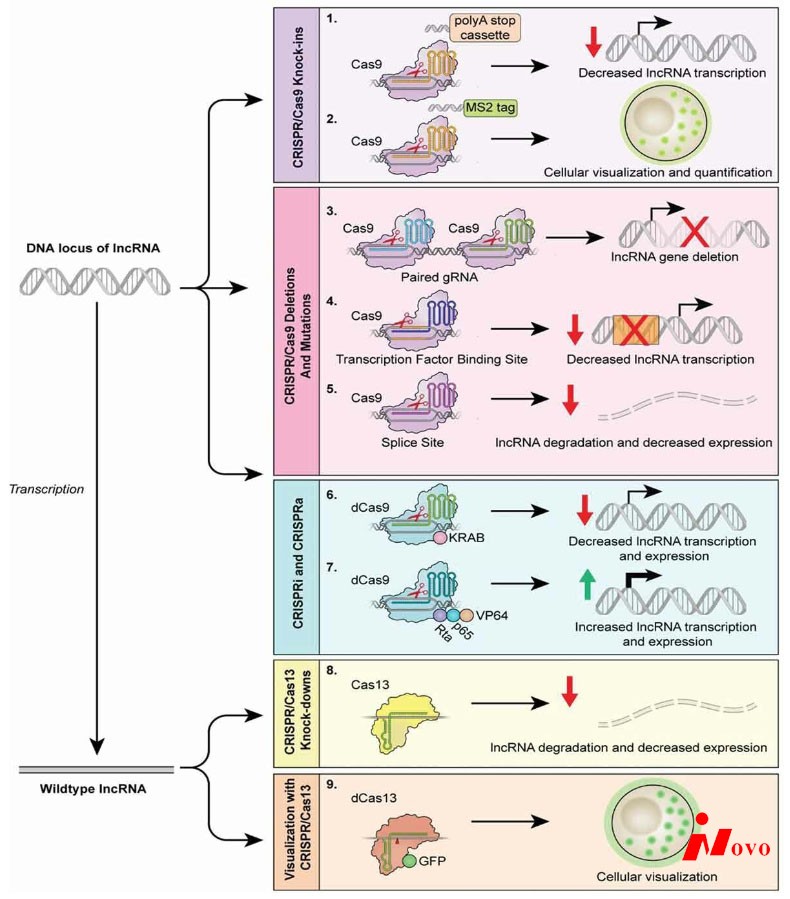
除了这些令人难以置信的机会,CRISPR系统还提供了进一步了解lnc RNA的机会,随着进一步的研究和开发,CRISPR系统理论上可以在临床上用于在治疗背景下操纵lnc RNA。CRISPR在小鼠体内纠正了点突变,并在体外用于治疗癌症,这表明CRISPR在未来可以用于体内治疗疾病[citation 97–99]。因为lncRNAs表达水平低,而且通常具有组织和细胞特异性,所以它们可能是治疗靶点的良好选择。当考虑CRISPR用于患者时,主要关注的是脱靶效应,尽管与蛋白质编码基因相比,当靶向lncRNA时,这可能不是一个问题。除了这种严重的担忧,由于缺乏物种间的保守性,研究lncRNAs的体内靶向性是复杂的。最后,使用CRISPR治疗性靶向lncRNAs的前景确实存在伦理问题,需要在临床使用该技术之前解决。从操纵传统的Cas9敲除到利用CRISPR-Disp等创新方法,CRISPR/Cas系统为关于lncRNAs功能的创新研究提供了无限的机会。

